

Alzheimer’s Dementia is physiologically characterized as a list of things: hyperphosphorylation of tau proteins, and the buildup of hard-to-rid-of beta-amyloid peptide fragments, especially Aβ42 (an extremely easily clumped species) and oligomers. Creation of Aβ42 is pathologically caused by the cleavage of amyloid precursor proteins (APP) by β-secretase enzymes outside of the Aβ region of the protein. Normally, when α-secretase cleaves this protein, it “destroys” this “stickier” region. However, β-secretase cleaves amyloid precursor proteins outside of this “stickier” region, essentially keeping it intact. After cleavage, γ-secretase is the enzyme that is responsible for “releasing” the cleaved fragments into the extracellular space. For a generalized understanding, the production of these isn’t the main problem in AD. It’s that the enzymes, which are responsible for “ridding” of them, are expressed in a lesser frequency, or are broken down due to oxidative stress. In other words, there is so much radioactive waste that your local garbage facility is starting to malfunction.
Here is a picture for better understanding:
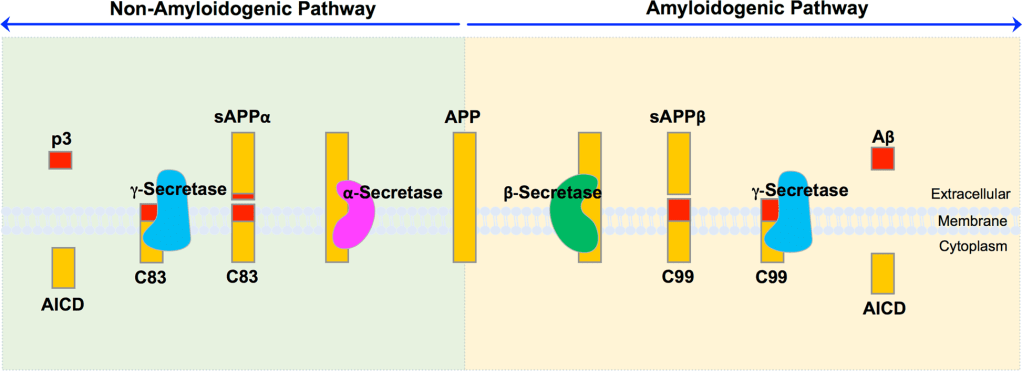
Hur, J.-Y. (2022). γ-Secretase in Alzheimer’s disease. Experimental & Molecular Medicine, 54(4), 433–446. https://doi.org/10.1038/s12276-022-00754-8
*Note (intricacies): The main difference between these pathways is in the location of the initial cleavage. The Aβ region spans both the extracellular domain and the transmembrane domain of APP. There is a portion “poking out” of the cell. α-secretase cleaves within this region, irreversibly disrupting the hydrophobic (sticky) Aβ sequence. In contrast, β-secretase cleaves immediately N-terminal to the Aβ region, leaving the full sequence intact. γ-secretase then performs intramembrane cleavage near the extracellular boundary, defining the length of Aβ (e.g., Aβ40 vs. Aβ42) and releasing the peptide into the extracellular space.
Tau
Phosphorylation is normally used for protein regulation; one could even call it a generalized protein mechanic and on/off switch, 2 in 1. Protein kinases add phosphate groups onto specific amino acids that make up the protein. Tau is an essential protein related to supporting the microtubule structures within the cell. Think of microtubules as the “highway” of a cell, essential for the transportation of vesicles and molecules. In the case of AD, the excessive addition of these phosphate groups leads to a “deformed” structure and detachment from the microtubules, forcing them to malfunction and form neurofibrillary tangles where, just like the beta-amyloid plaques, a build-up of these malfunctioning pieces starts to prove detrimental to neural cells.
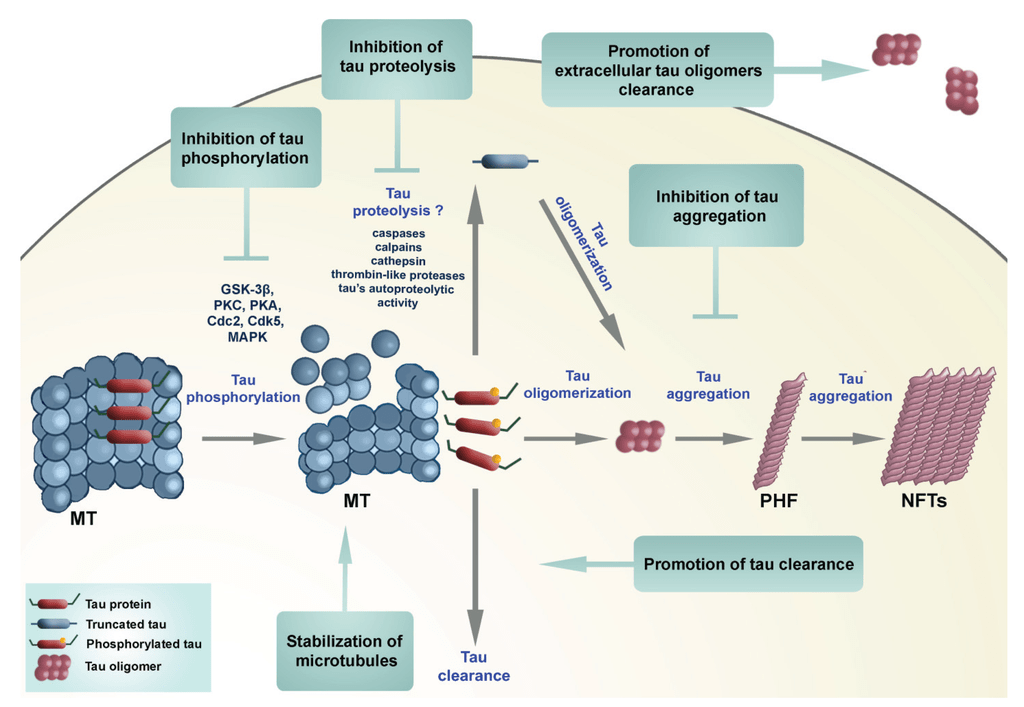
Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. https://doi.org/10.3390/biom6010006
*Note about tau phosphorylation (intricacies): Under physiological conditions, tau binds and stabilizes neuronal microtubules (MT). In disease states, tau becomes abnormally phosphorylated by kinases (e.g.,GSK-3β, Cdk5, MAPK, which are the more “reputable” ones), reducing MT binding and increasing the pool of soluble tau. Detached tau may then go through proteolysis to generate products that promote misfolding, then self-associate into toxic tau oligomers. They are small, soluble clusters made of a few tau molecules (intermediate assemblies that are larger than single tau monomers but not yet formed into long fibrils). These oligomers can then aggregate into paired helical filaments (PHFs) and then finally neurofibrillary tangles (NFTs).
Fun Fact!: One of the most accurate modern biomarkers for Alzheimer’s disease is p-tau217. It’s a specific phosphorylated form of tau. Levels of p-tau217 in blood or cerebrospinal fluid can lead to early detection and diagnosis of Alzheimer’s dementia.
Aβ and Tau “Coupling”
The pathologies for amyloid-β and tau hyperphosphorylation can seem like two different ones with no correlation to one another on the surface level. However, that is not the case. As a precursor to the bigger amyloidogenic plaques formed, the soluble amyloid-β oligomers are toxic to the synapses, disrupting fundamental neural properties such as calcium ion balances and excitability. These, in turn, cause the activation of more kinases that phosphorylate tau, such as MAPK, GSK-3β, and CDK5. This then leads to a very obvious consequence: the increase in delocalized aggregate-prone tau fragments.
It has been more and more clear that amyloid pathology alone causes limited neurodegeneration, whereas the tau pathology, on the other hand, leads to a sudden increase in observable degeneration. Within this logic map, amyloid-β oligomers and plaques act as accelerators of tau pathology, while tau dysfunction itself serves as the “smoking gun”. One could say they are basically “coupled”.
Mitochondrial Dysfunction
Alzheimer’s disease is not only characterized by plaques and tangles but also by an energy failure within neurons. Neurons rely heavily on mitochondria to produce ATP and to regulate cellular stress. In Alzheimer’s disease, mitochondrial efficiency declines, leading to reduced energy production and increased generation of damaging byproducts such as reactive oxygen species. Because synapses have exceptionally high energy demands, this dysfunction often manifests first as impaired communication between neurons rather than immediate cell death. In fact, it is a known fact that up to 80% of the energy used in the brain is due to synaptic functions. Importantly, mitochondrial dysfunction is closely intertwined with the classical pathological features of Alzheimer’s disease. Amyloid beta can directly stress mitochondria and disrupt calcium homeostasis, while pathological tau destabilizes microtubules and interferes with axonal transport, preventing healthy mitochondria from reaching synapses. Together, these processes worsen the other and accelerate neurodegeneration.
Oxidative Stress
The problem of oxidative stress in AD is largely due to the generation and lack of protection from reactive oxygen species (ROS), which are created when electrons get “extracted” prematurely on their journey down the electron transport chain (ETC). They are supposed to end up at Complex IV (AKA cytochrome c oxidase), but sometimes an electron escapes early, most often from Complex I or III. As a result, ROSs form, which have extremely high affinities for nearby electrons due to their “unstable” electron configurations. These, in turn, can damage essential building blocks of cellular function, including mitochondrial DNA and cell membranes.
*Note about ROS formation and impact (intricacies): Superoxide (O₂⁻) is the first “ROS” made directly from the electron “leakage”. The cell then, catalyzed by superoxide dismutase (SOD), combines it with 2 hydrogens to form hydrogen peroxide (H₂O₂). Hydrogen peroxide isn’t dangerous by nature; what makes it so unpleasant is the fact that it will readily convert to a radical species when it is near copper or iron. Unfortunately, our brain is a HEAVILY iron/copper-concentrated region. This reaction is also known as the Fenton reaction. DNA is damaged because ROSs react with bases and the sugar phosphate groups, especially guanine. Thus, this can cause breakage in single and double strands, which in essence are shared electrons between specific atoms. Membranes are damaged because a large part of the cellular membranes consists of polyunsaturated fatty acids. Unsaturated fatty acids consist of at least one double bond in their structure, which act as “weak points” also known as allylic hydrogens that are easier to steal. The radical steals the hydrogen from said fatty acids, converting it into a radical. The radicalized fatty acid then reacts with oxygen, forming a lipid peroxyl radical, which steals hydrogen from a neighboring fatty acid, essentially starting a “wildfire”.
Cerebrovascular Dysfunction and Hypoperfusion
Alzheimer’s disease is also associated with widespread vascular dysfunction in the brain. Neurons depend on a continuous supply of oxygen and glucose delivered through cerebral blood flow to sustain mitochondrial energy production. In Alzheimer’s disease, this supply becomes compromised due to impaired neurovascular coupling and reduced perfusion, leading to chronic, low level hypoxia and metabolic stress. Because neuronal energy demand remains high even at rest, reduced blood flow places persistent strain on mitochondria and accelerates synaptic failure. Importantly, cerebrovascular dysfunction does not occur in isolation. It interacts with amyloid and tau pathology, oxidative stress, and mitochondrial impairment, forming a convergence point where multiple pathological processes amplify one another.

Leave a comment